Tau Silences, Aβ Inflames; Hitting Excitatory Synapses Hardest
Quick Links
Part 2 of a two-part story. Click here for Part 1.
Long before neurons die in the Alzheimer’s brain, their synapses go kaput. Both Aβ and tau pathology have been blamed for this synaptic breakdown, but at the Society for Neuroscience meeting, held November 3–7 in San Diego, researchers dissected how putting the two culprits together wields a blow quite distinct from either alone. They found that tau silenced neuronal activity, while Aβ stoked it. When placed together in the same mouse, tau’s silencing power won out. Curiously, tau turned down synaptic genes that operate specifically in excitatory neurons. And a new paper reported that a distinct gene-expression signature in excitatory neurons might explain why these cells are highly vulnerable to tau aggregation, as opposed to the relatively tau-resistant inhibitory neurons or glial cells.
- Aβ stokes neuronal activity; tau silences it.
- When expressed together, tau wins out.
- Tau pathology downregulates expression of synaptic genes.
- Only excitatory neurons express genes that promote tau aggregation.
At the Synapse, Do Aβ and Tau Clash or Conspire?
Both Aβ and tau aggregates have been blamed for synaptic dysfunction. What happens when the two meet? Two complementary lines of evidence presented at SfN suggest an answer more complex than simple synergy. Tau accumulation has been spotted in both pre-and postsynaptic compartments in autopsy brains, and also in mouse models of neurodegeneration. However, the field lacks models that illuminate the combined synaptic deficits evoked by tau and the other cardinal AD pathology, Aβ. To make one, researchers led by Marc Aurel Busche and Bradley Hyman of Massachusetts General Hospital in Boston crossed rTg21221 or rTg4510 mice, which express the wild-type or P301L mutated form of human tau, respectively, with APP/PS1 mice, which carry familial AD mutations in APP and PS1. They then asked the question, how do tau and Aβ interact to affect neuronal activity?
At SfN, Simon Dujardin first described analyses in the single strains. Measuring calcium transients related to spontaneous action potentials in cortical neurons, the researchers observed a distinct uptick in neuronal activity in APP/PS1 mice compared with wild-type controls. The opposite was true in rTg21221 mice, where neuronal firing was suppressed, and a large fraction of neurons appeared silent. Neurons were similarly shushed in rTg4510 mice, which develop neurofibrillary tangles owing to their expression of the aggregation-prone P301L mutant. These findings suggested that oligomeric forms of tau, as found in both the rTg21221 and rTg4510 strains, rather than NFTs, were responsible for the synaptic deficits observed in these mouse strains.
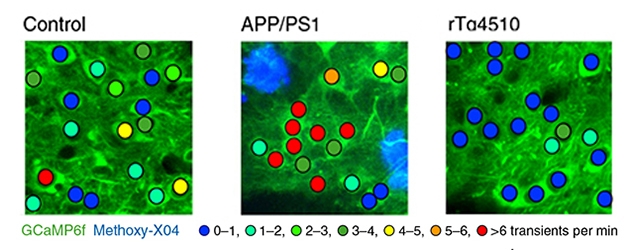
Loud Aβ, Quiet Tau. Neuronal activity was measured by the frequency of calcium transients in parietal cortices of control, APP/PS1, or rTg4510 mice. Neurons in APP/PS1 mice were hyperactive (red), while rTg4510 were silent (blue). [Courtesy of Busche et al., Nature Neuroscience, 2018.]
What would happen in the crosses? Dujardin reported that the neuronal hyperactivity in APP/PS1 mice was no match for the silencing power of tau. Regardless of whether the APP/PS1 were crossed to rTg21221 or to the tangle-bearing rTg4510 mice, neuronal firing in the crosses’ cortices was doused. Things got strange when the researchers turned off tau expression in six-month-old mice. After six weeks without transgenic human tau, the researchers were surprised to find that neurons were still hushed on the APP/PS1 background. However, in rTg21221 or rTg4510 mice, turning off tau was sufficient to return neuronal firing to normal. The researchers obtained similar results when they turned off tau earlier, in three- to four-month-old mice. They concluded that something about the presence of Aβ prevented the neurons from regaining their activity after tau was turned down. They hypothesized that when faced with both Aβ and tau, struggling synapses were less apt to recover.
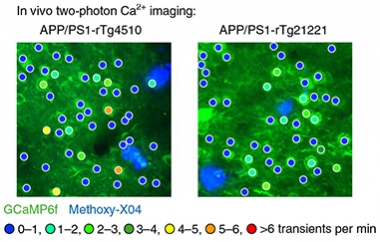
Overpowered by Silence.
Neurons in APP/PS1 animals crossed to either tau strain were relatively silent, compared with control or APP/PS1 mice (compare with previous figure). [Courtesy of Busche et al., Nature Neuroscience, 2018.]
The results appeared December 17 in Nature Neuroscience. The findings are based on overexpression models, but even so, Busche and colleagues write that therapeutic strategies aimed at either only Aβ or only tau might be confounded by the complex relationship between the two. Busche recently started a lab at University College London.
At SfN, Tara Spires-Jones of the University of Edinburgh, U.K., described her results from making similar crosses of tau and Aβ mice. Like Busche, Spires-Jones generated rTg21221 mice crossed to APP/PS1 animals. As opposed to a previous cross Spires-Jones reported, all the strains in her current study were generated on a tau knockout background to avoid confounds with endogenous mouse tau (Jackson et al., 2016). Spires-Jones compared AD pathology, gene expression, markers of synaptic loss, and behavioral deficits among the mouse strains. Compared with APP/PS1 mice, the APP/PS1 x rTg21221 offspring (a.k.a. MAPT-AD) had fewer, smaller plaques. Neither developed tau tangles, but the MAPT-AD mice had marked accumulation of tau in both pre- and postsynaptic compartments. Curiously, the MAPT-AD mice were hyperactive, running around more than control mice in open spaces.
Comparing the whole-brain transcriptomes of MAPT-AD, APP/PS1, rTg21221, and tau-knockout control strains, Spires-Jones reported a surge in expression of genes involved in inflammation, such as Trem2, Gfap, Cd68, and C1q, in both the APP/PS1 mice and MAPT-AD mice, over tau knockout controls or rTg21221 mice. Gene expression was no different between rTg21221 and controls. Though most of the pro-inflammatory gene-expression signature in MAPT-AD mice appeared in the APP/PS1 animals as well, a few inflammatory genes only revved up in the crosses, including Lilrb4, Ccl3, and Cst7.
The gene encoding cellular prion protein, PrPc, was strongly upregulated in the crosses. This drew the researchers’ attention because Aβ and PrPc reportedly interact at synapses, damaging them (Jul 2012 news). Strikingly, Spires-Jones also reported reduced expression of genes involved in synaptic function over each parental strain, suggesting that tau and Aβ pathology cooperatively turned them down. Spires-Jones speculated that this tau-induced decrease in transcripts encoding key synaptic genes could explain Busche’s data showing that tau dampens neuronal activity. When Spires-Jones switched off tau expression in 10-month-old MAPT-AD mice with doxycycline, these synaptic gene transcripts rose back to normal. Additionally, turning off tau relieved the animals of their hyperactivity.
However, turning off tau did not bring back synapses. Compared to tau knockout control or rTg21221 mice, both APP/PS1 mice and MAPT-AD mice had a third fewer synapses, especially near plaques, and they did not return after tau was switched off. Interestingly, Spires-Jones noted that rarely did Aβ and tau congregate in the same pre- or postsynaptic compartments. Aβ oligomers associated with synapses next to plaques, but tau accumulated equally in synapses near and far from plaques. The data is posted on bioRχiv.
“The impact of pathology on networks is of significant interest, and the use of models that have both amyloid and tau pathology, which better represent human AD, are a move in the right direction,” commented Karen Duff of Columbia University Medical Center, New York. “We still need to move toward more physiological levels of the proteins, with the correct spatial and temporal expression patterns to properly represent regional and cell-type vulnerability, but the interplay between the two pathologies is interesting,” she added.
In the manuscript, the scientists offer several explanations for synaptic loss orchestrated by Aβ and tau. It could be due to the inflammatory response, to association between Aβ and PrPc and, lastly, the downregulation of synaptic genes. Spires-Jones said that all downregulated synaptic genes—including AMPA and NMDA receptor subunits Gria2, Gria3, Gria4, Grin2a, Homer2, and Camk2b—function in excitatory, not inhibitory, neurons. This is in keeping with a recent study that reported a selective vulnerability of excitatory neurons to tau pathology (Jan 2017 news). Duff, Hongjun (Harry) Fu, and Abid Hussaini, also at Columbia, saw tau pathology accumulate in excitatory, not inhibitory, entorhinal cortex neurons of mice expressing P301L-tau. Excitatory cells selectively perished, dropping by as much as 70 percent in number compared with controls, while inhibitory neurons thrived.
A new study, led by Duff, Fu (now at Ohio State University), and Michele Vendruscolo at the University of Cambridge, U.K., provides a potential explanation for the excitatory neurons’ vulnerability. In the December 17 Nature Neuroscience, the scientists report that excitatory neurons in mouse and human brains express a cadre of genes known to coax tau into aggregation.

Tau’s Tangled Web. Groups of co-expressed genes associated with protein aggregation (red), tau tangles (green), aggregation protectors (blue) and promoters (yellow, upper left), tau (black), and genes shared between MS and tangles (brown). [Courtesy of Fu et al., Nature Neuroscience, 2018.]
The researchers extended the mouse findings linking tau accumulation to excitatory neurons to the human brain. They reported that in postmortem brain samples from AD patients compared with controls, tau pathology co-localized with markers of excitatory neurons, not inhibitory ones. This was true in the entorhinal cortex, where tau pathology starts, as well as in the surrounding neocortex, where tau spreads later in disease.
Why this selective vulnerability? To investigate, the researchers turned to two independent single-nucleus RNA-sequencing data sets—SNS and DroNc-Seq (Lake et al., 2016; Habib et al., 2017). These gene-expression data sets were generated by sequencing RNA from thousands of single nuclei. The nuclei were sorted from postmortem brain tissue of people who died without AD or any other neuropathology.
The researchers at Cambridge, led by Vendruscolo, found that nuclei hailing from excitatory neurons had a gene-expression signature distinctive from those derived from inhibitory neurons. In particular, the researchers identified opposing patterns of expression in a suite of previously identified genes making up a “protein homeostasis signature” (Aug 2016 news). Excitatory neurons had an overabundance of transcripts encoding proteins known to co-aggregate with tau or to promote tau aggregation. Conversely, they had a dearth of transcripts encoding genes that protect against tau aggregation. Nuclei from inhibitory neurons, or glial cells, had the opposite pattern.
In a co-expression network analysis, Fu and colleagues zeroed in on BAG3. This autophagy-related chaperone appears to orchestrate the aggregation protection network in inhibitory neurons (Behl, 2011). Working with Gail Johnson and Maoping Tang at the University of Rochester in New York, the researchers found elevated BAG3 protein levels in inhibitory compared with excitatory neurons in postmortem brain samples from people with and without AD. Further, modulating BAG3 expression in primary cultured neurons affected tau accumulation. BAG3 overexpression prevented tau accumulation, while knocking it down promoted a tau buildup. Duff said the researchers are now interested in why excitatory neurons are selectively vulnerable to tau homeostasis pathways and how that leads to tangle formation.
Spires-Jones commented that all three studies converge on the idea that pathological tau impairs excitatory neuron function. “The findings of Karen Duff and colleagues showing that excitatory neurons have a propensity to accumulate tau due to their expression patterns of genes involved in protein homeostasis are very interesting, and help explain why we observe accumulation of small tau aggregates in excitatory synapses both in our mouse model and in human AD brain,” she added. Spires-Jones also pointed out that while BAG3 levels were not changed in the MAPT-AD mice, transcripts encoding BAG5, a related protein, did drop.—Jessica Shugart
References
News Citations
- Toxic Tau: Who Are You, and Where Are You From?
- Tracing Aβ’s Toxicity Through Prion Protein, Fyn Kinase
- Led Astray: Pathology Tied to “Grid Cell” Malfunction in Tauopathy Model
- Aggregation-Prone Gene Expression Signature Mapped in Brain
Research Models Citations
Paper Citations
- Jackson RJ, Rudinskiy N, Herrmann AG, Croft S, Kim JM, Petrova V, Ramos-Rodriguez JJ, Pitstick R, Wegmann S, Garcia-Alloza M, Carlson GA, Hyman BT, Spires-Jones TL. Human tau increases amyloid β plaque size but not amyloid β-mediated synapse loss in a novel mouse model of Alzheimer's disease. Eur J Neurosci. 2016 Dec;44(12):3056-3066. Epub 2016 Nov 12 PubMed.
- Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S, Vijayaraghavan R, Wong J, Chen A, Sheng X, Kaper F, Shen R, Ronaghi M, Fan JB, Wang W, Chun J, Zhang K. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016 Jun 24;352(6293):1586-90. PubMed.
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, Weitz DA, Rozenblatt-Rosen O, Zhang F, Regev A. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods. 2017 Oct;14(10):955-958. Epub 2017 Aug 28 PubMed.
- Behl C. BAG3 and friends: co-chaperones in selective autophagy during aging and disease. Autophagy. 2011 Jul;7(7):795-8. PubMed.
External Citations
Further Reading
Primary Papers
- Busche MA, Wegmann S, Dujardin S, Commins C, Schiantarelli J, Klickstein N, Kamath TV, Carlson GA, Nelken I, Hyman BT. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat Neurosci. 2019 Jan;22(1):57-64. Epub 2018 Dec 17 PubMed.
- Fu H, Possenti A, Freer R, Nakano Y, Hernandez Villegas NC, Tang M, Cauhy PV, Lassus BA, Chen S, Fowler SL, Figueroa HY, Huey ED, Johnson GV, Vendruscolo M, Duff KE. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat Neurosci. 2019 Jan;22(1):47-56. Epub 2018 Dec 17 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.